



Research Articles
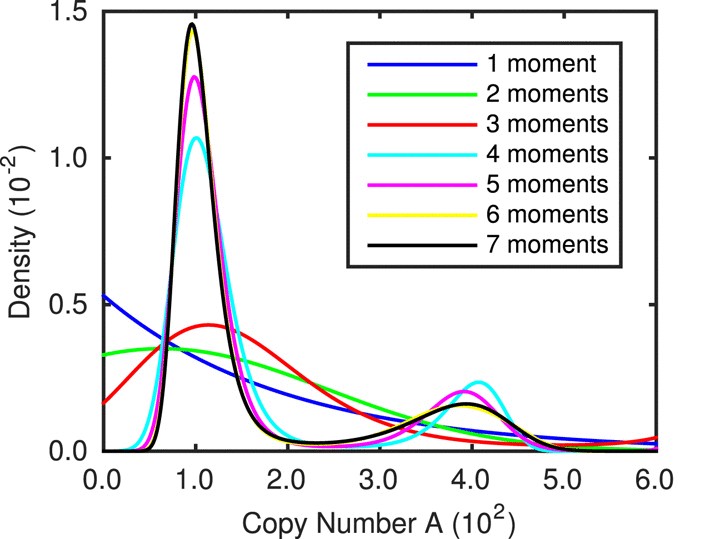
Abstract: Biochemical reaction networks are often modelled using discrete-state, continuous-time Markov chains. System statistics of these Markov chains usually cannot be calculated analytically and therefore estimates must be generated via simulation techniques. There is a well documented class of simulation techniques known as exact stochastic simulation algorithms, an example of which is Gillespie’s direct method. These algorithms often come with high computational costs, therefore approximate stochastic simulation algorithms such as the tau-leap method are used. However, in order to minimise the bias in the estimates generated using them, a relatively small value of tau is needed, rendering the computational costs comparable to Gillespie’s direct method. The multi-level Monte Carlo method (Anderson and Higham, Multiscale Model. Simul. 10:146–179, 2012) provides a reduction in computational costs whilst minimising or even eliminating the bias in the estimates of system statistics. This is achieved by first crudely approximating required statistics with many sample paths of low accuracy. Then correction terms are added until a required level of accuracy is reached. Recent literature has primarily focussed on implementing the multi-level method efficiently to estimate a single system statistic. However, it is clearly also of interest to be able to approximate entire probability distributions of species counts. We present two novel methods that combine known techniques for distribution reconstruction with the multi-level method. We demonstrate the potential of our methods using a number of examples.
Authors: D. Wilson and R. E. Baker
Link to article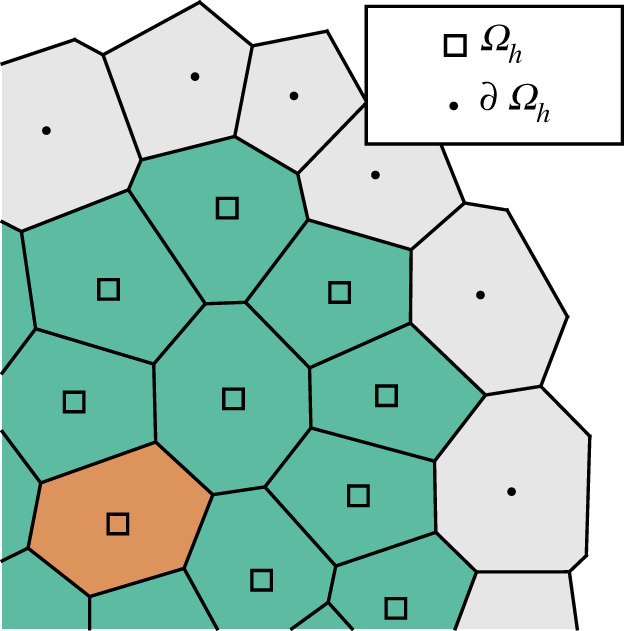
Abstract: The processes taking place inside the living cell are now understood to the point where predictive computational models can be used to gain detailed understanding of important biological phenomena. A key challenge is to extrapolate this detailed knowledge of the individual cell to be able to explain at the population level how cells interact and respond with each other and their environment. In particular, the goal is to understand how organisms develop, maintain and repair functional tissues and organs. In this paper, we propose a novel computational framework for modelling populations of interacting cells. Our framework incorporates mechanistic, constitutive descriptions of biomechanical properties of the cell population, and uses a coarse-graining approach to derive individual rate laws that enable propagation of the population through time. Thanks to its multiscale nature, the resulting simulation algorithm is extremely scalable and highly efficient. As highlighted in our computational examples, the framework is also very flexible and may straightforwardly be coupled with continuous-time descriptions of biochemical signalling within, and between, individual cells.
Authors: S. Engblom, D. B. Wilson and R. E. Baker
Link to article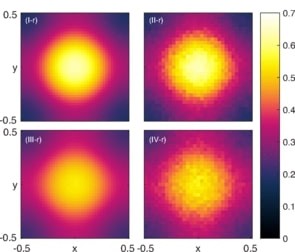
Abstract: Complex biological and physical transport processes are often described through systems of interacting particles. The effect of excluded volume on these transport processes has been well studied; however, the interplay between volume exclusion and reactions between heterogenous particles is less well studied. In this paper we develop a framework for modeling reaction-diffusion processes which directly incorporates volume exclusion. We consider simple reactions (unimolecular and bimolecular) that conserve the total number of particles. From an off-lattice microscopic individual-based model we use the Fokker-Planck equation and the method of matched asymptotic expansions to derive a low-dimensional macroscopic system of nonlinear partial differential equations describing the evolution of the particles. A biologically motivated, hybrid model of chemotaxis with volume exclusion is explored, where reactions occur at rates dependent upon the chemotactic environment. Further, we show that for reactions that require particle contact the appropriate reaction term in the macroscopic model is of lower order in the asymptotic expansion than the nonlinear diffusion term. However, we find that the next reaction term in the expansion is needed to ensure good agreement with simulations of the microscopic model. Our macroscopic model allows for more direct parametrization to experimental data than existing models.
Authors: D. B. Wilson, H. Byrne and M. Bruna
Link to article
Abstract: This work investigates multi-resolution methodologies for simulating dimer models. The solvent particles which make up the heat bath interact with the monomers of the dimer either through direct collisions (short-range) or through harmonic springs (long-range). Two types ofmulti-resolution methodologies are considered in detail: (a) describing parts of the solvent far away from the dimer by a coarser approach; (b) describing each monomer of the dimer by using a model with different level of resolution. These methodologies are then used to investigate the effect of a shared heat bath versus two uncoupled heat baths, one foreachmonomer. Furthermore, the validity of themulti-resolutionmethods is discussed by comparison to dynamics of macroscopic Langevin equations.
Authors: R. Gunaratne, D. B. Wilson, M. Flegg, R. Erban
Link to article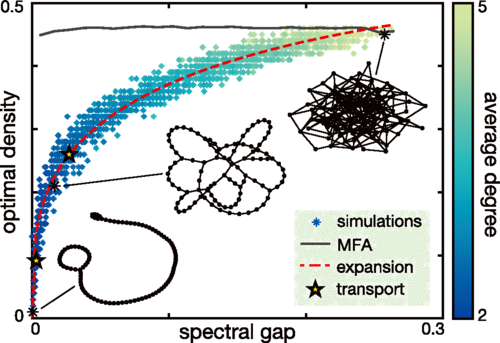
Abstract: A random search process in a networked environment is governed by the time it takes to visit every node, termed the cover time. Often, a networked process does not proceed in isolation but competes with many instances of itself within the same environment. A key unanswered question is how to optimize this process: How many concurrent searchers can a topology support before the benefits of parallelism are outweighed by competition for space? Here, we introduce the searcher-averaged parallel cover time (APCT) to quantify these economies of scale. We show that the APCT of the networked symmetric exclusion process is optimized at a searcher density that is well predicted by the spectral gap. Furthermore, we find that nonequilibrium processes, realized through the addition of bias, can support significantly increased density optima. Our results suggest alternative hybrid strategies of serial and parallel search for efficient information gathering in social interaction and biological transport networks.
Authors: D. B. Wilson, R. E. Baker and F. G. Woodhouse
Link to article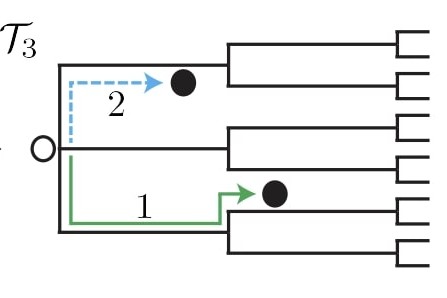
Abstract: Consider a particle whose position evolves along the edges of a network. One definition for the displacement of a particle is the length of the shortest path on the network between the current and initial positions of the particle. Such a definition fails to incorporate information of the actual path the particle traversed. In this work we consider another definition for the displacement of a particle on networked topologies. Using this definition, which we term the winding distance, we demonstrate that for Brownian particles, confinement to a network can induce a transition in the mean squared displacement from diffusive to ballistic behavior for long times. A multiple scales approach is used to derive a macroscopic evolution equation for the displacement of a particle and uncover a topological condition for whether this transition in the mean squared displacement will occur. Furthermore, for networks satisfying this topological condition, we identify a prediction of the timescale upon which the displacement transitions to long-time behavior. Finally, we extend the investigation of displacement on networks to a class of anomalously diffusive transport processes, where we find that the mean squared displacement at long times is affected by both network topology and the character of the transport process.
Authors: D. B. Wilson, R. E. Baker and F. G. Woodhouse
Link to article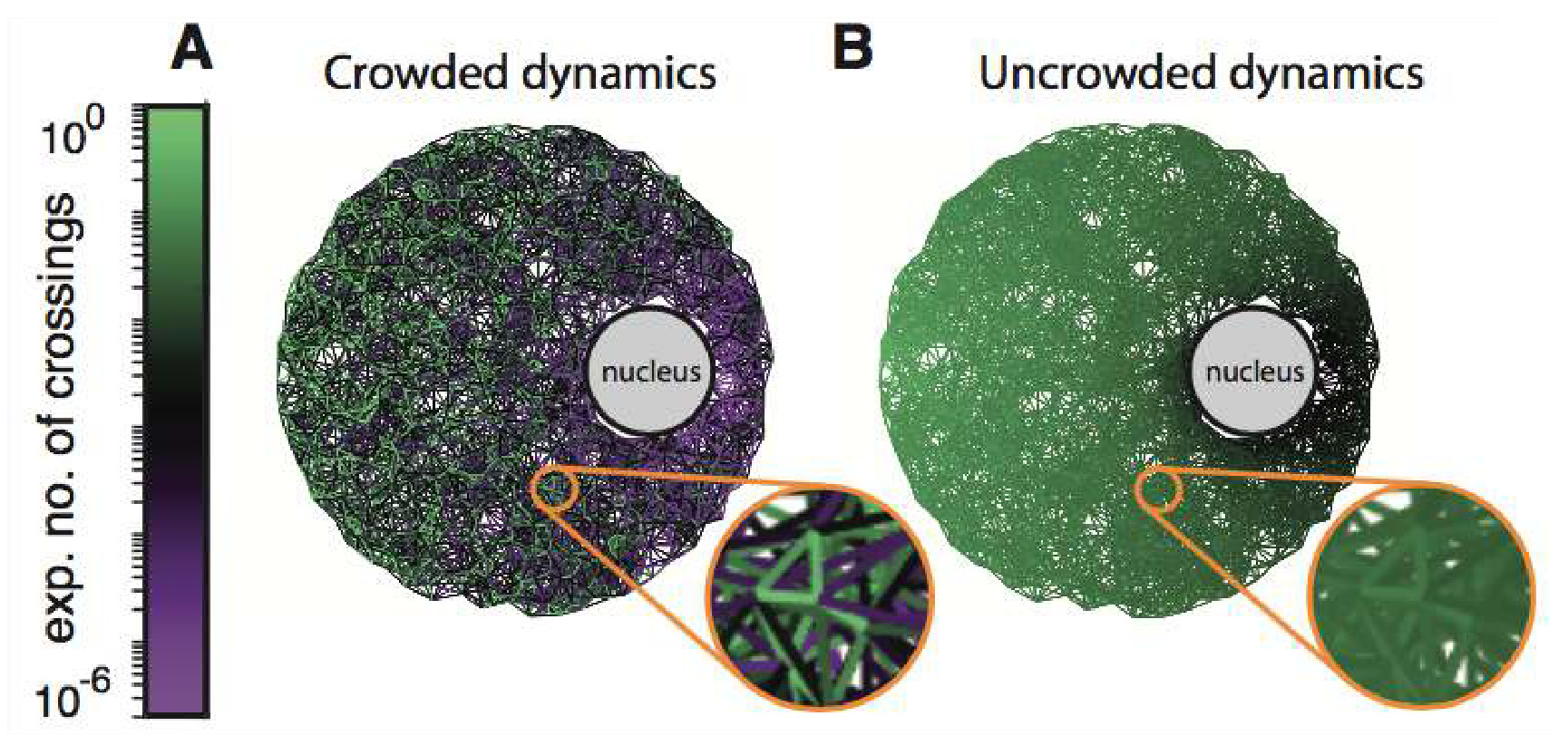
Abstract: Transport phenomena within crowded, complex environments is observed at many spatial and temporal scales, from pedestrian traffic in cities and buildings to macromolecular motion within cells and ion exchange in batteries. Geometric restrictions in an environment can hinder the motion of individuals and, combined with crowding between the individuals, can have drastic effects on global transport behaviour. However, in general, the interplay between crowding and geometry is poorly understood. Existing techniques to predict the behaviour of crowded transport processes approximate complex environments as high-dimensional meshes and use computationally expensive models that lack the ability to reveal the functional influence of geometry and crowding on transport. Here, we employ networked representations of complex environments and provide an efficient, foundational framework within which the combined roles of geometry and crowding can be explored. Multiple models of crowded, networked transport are derived that are capable of extracting detailed information at both the level of the whole population or an individual within it. A combination of theoretical and numerical analysis identifies critical topological features of environments that enable accurate prediction of temporal and spatial transport statistics, as well as insight into the design of optimal networks. Our approach is applicable to transport processes across a broad range of scientific disciplines, bypasses traditional computational challenges of discretisation, and establishes a unified connection between geometry, crowding and transport.
Authors: D. B. Wilson, F. G. Woodhouse, M. J. Simpson and R. E. Baker
Link to article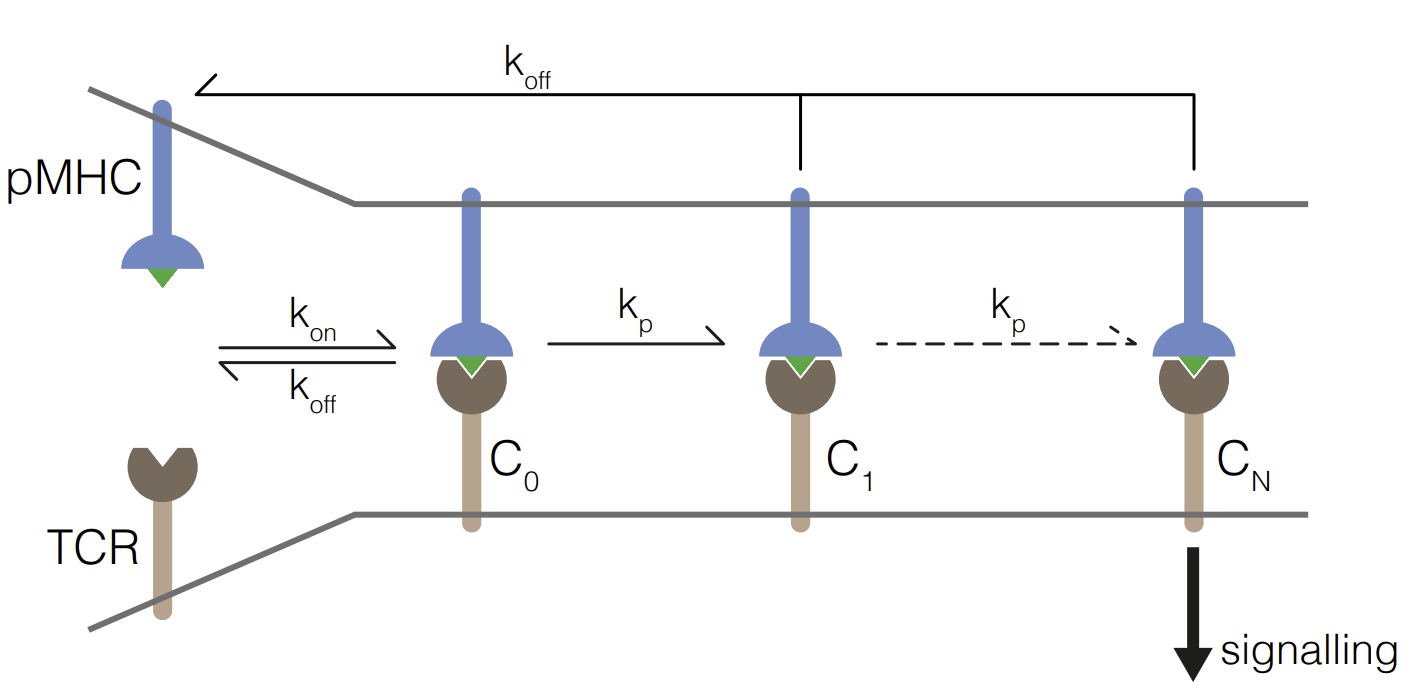
Abstract: T cells use their T cell receptors (TCRs) to discriminate between peptide MHC (pMHC) ligands that bind with different affinities but precisely how different remains controversial. This is partly because the affinities of physiologically relevant interactions are often too weak to measure. Here, we introduce a surface plasmon resonance protocol to measure ultra-low TCR/pMHC affinities (KD~1000μM). Using naïve, memory, and blasted human CD8+ T cells we find that their discrimination power is unexpectedly low, in that they require a large >100-fold decrease in affinity to abolish responses. Interestingly, the discrimination power reduces further when antigen is presented in isolation on artificial surfaces but can be partially restored by adding ligands to CD2 or LFA-1. We were able to fit the kinetic proof-reading model to our data, yielding the first estimates for both the time delay (2.8 s) and number of biochemical steps (2.67). The fractional number of steps suggest that one of the proof-reading steps is not easily reversible.
Authors: J. Pettmann, E. Abu-Shah, M. Kutuzov, D. B. Wilson, M. L. Dustin, S. J. Davis, P. Anton van der Merwe and O. Dushek
Link to article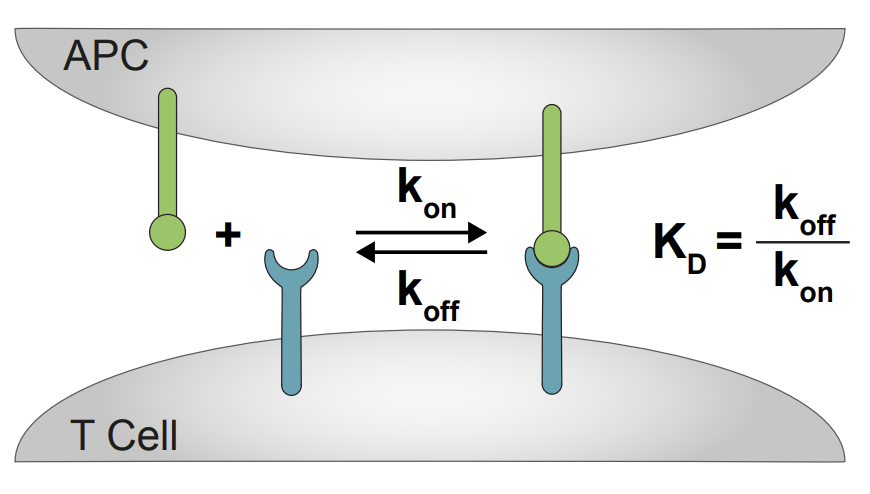
Abstract: T cells use their T-cell receptors (TCRs) to discriminate between lower-affinity self and higher-affinity non-self pMHC antigens. The strength of this discrimination and the mechanisms that produce it remain controversial. Although a large number of mouse and human TCRs have now been characterised, they have not been used to precisely quantitate discrimination. Here, we systematically quantify the discrimination of TCRs using a discrimination power (α). Early influential studies on three mouse TCRs suggested that discrimination was nearly perfect (α ~9.0). In striking contrast, our analysis of published data on other mouse and human TCRs, and more recent data on the original mouse TCRs, produced significantly lower discrimination (α = 2.0). Although not perfect, the discriminatory power of TCR was greater than that of conventional receptors such as cytokine receptors, GPCRs, RTKs, and CARs (α≤1). The revised discriminatory power of the TCR is readily explained by a kinetic proof-reading mechanisms, and accounts for the ability of low affinity self-antigens to stimulate autoimmune and anti-tumour T cell responses.
Authors: A. Huhn, D. B. Wilson, P. Anton van der Merwe and O. Dushek
Link to article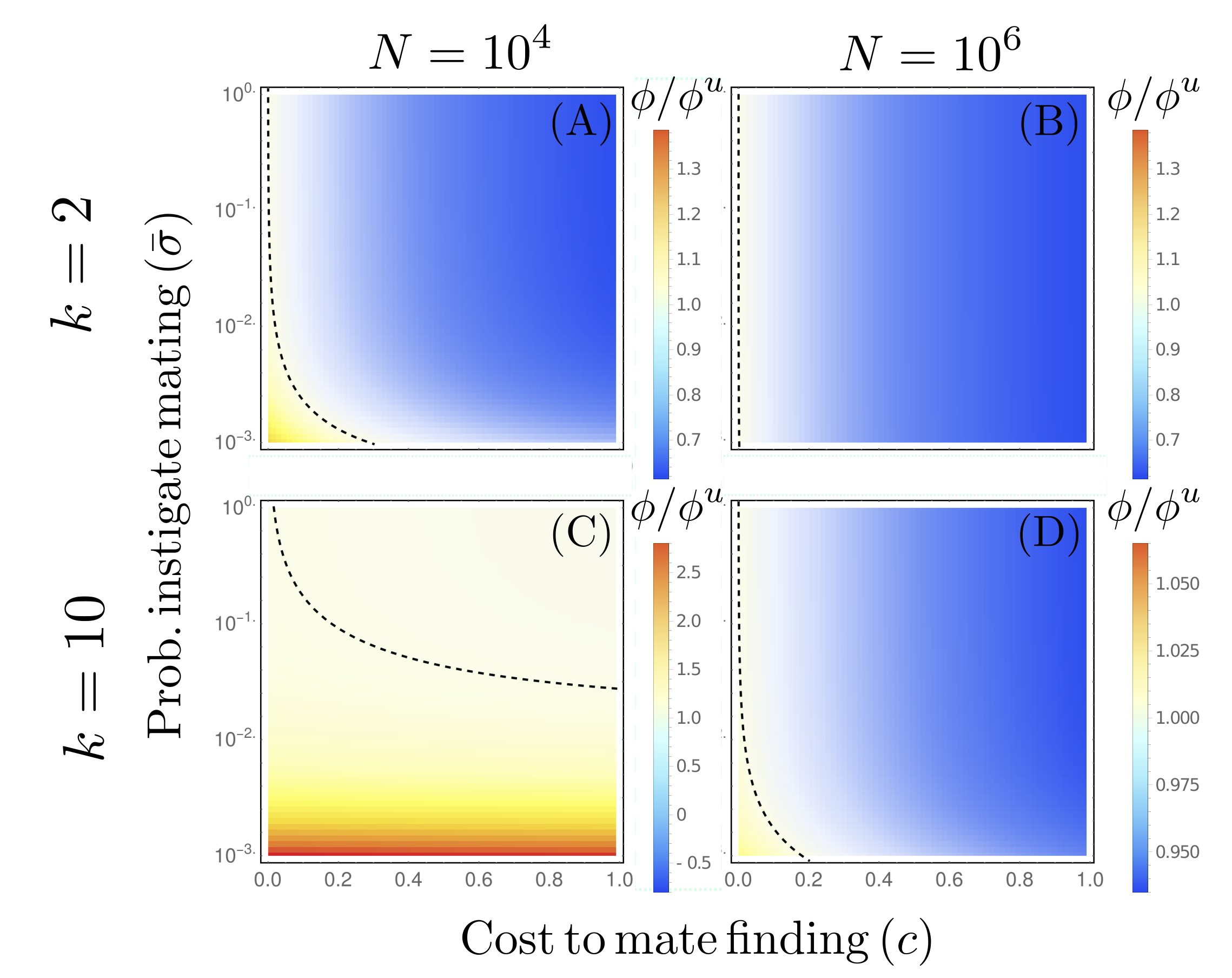
Abstract: The evolutionary mechanism that drove the establishment of self-incompatibility in early sexual eukaryotes is still a debated topic. While a number of competing hypotheses have been proposed, many have not received detailed theoretical attention. In particular, the hypothesis that self-incompatibility increases the benefits of genetic recombination in sexual haploids has been comparatively understudied. In this paper we address this topic by mathematically deriving how the probability of mating with a genetically distinct individual changes as a function of the presence or absence of self-incompatible mating type classes. We find that although populations with mating types successfully engage in sexual reproduction less frequently than their self-compatible competitors, they can nevertheless engage in useful sex with genetically distinct partners \emph{more} frequently. This conclusion holds when the number of sexual reproductive events per generation is low (i.e. in small populations with low rates of facultative sexual reproduction). Finally we demonstrate the potential for frequency-dependent selection in competitive dynamics between self-compatible and self-incompatible types. These analytic results provide a baseline for studying the sex advantage enhancer model for the evolutionary origin of mating types within each specific hypothesis for the evolution of recombination.
Authors: A.S.A. Smith, S. Pennington, I. Letter, D. B. Wilson and G. W. A. Constable
Link to article